Building a cleaner, healthier, and more sustainable world requires a better understanding of materials. Quantum simulation can help with the accurate and efficient calculation of the fundamental properties of materials. It can augment the ability of other simulation and experimental methods to predict the outcome of chemical reactions, such as catalytic reactions in materials science, enzymatic reactions in the life sciences, and electrochemical reactions for next-generation batteries.
Progress in accurate and efficient electronic structure simulation on classical (non-quantum) hardware has been tremendous in the last few decades. Quantum computers have the potential to further enhance our ability to perform such calculations. However, it is hard to say when this enhancement will happen—or what hardware characteristics will be needed for it to occur.
“Since the proposed framework can scale up with the rapidly evolving quantum technology, it can be used to significantly reduce qubit requirements for simulating … industrially relevant compounds, thereby extending the reach of quantum computation in the coming years.”
It is known, though, that in the near term, quantum computers will be of intermediate scale, composed of 10s–100s of qubits. Furthermore, quantum computation is prone to errors due to loss of quantum information, as quantum states are fragile and sensitive to various internal and external sources of noise. As a result, to assess and activate the potential of quantum computers for quantum simulations, the molecular system has to be represented—and errors must be mitigated—using few qubits.
What Can Be Done?
In a recent scientific paper,1 researchers at 1QBit and Dow proposed a new approach to achieve accurate and efficient electronic structure simulation on quantum computers. In collaboration with IonQ, the team demonstrated that this approach can achieve very high accuracy when deployed on a trapped-ion quantum computer.
In this work, it was demonstrated that the problem can be broken down into smaller pieces to reduce the number of qubits required to simulate the electronic structure of a large model chemical system. This allows for electron correlation effects to be more accurately accounted for—a hard-to-compute, but critically important, component of a molecule’s electronic structure.
The approach was applied to a ring of 10 hydrogen atoms, which is a common benchmark for evaluating electronic structure methods. High accuracy, known as chemical accuracy, was achieved when the results were compared to conventional full configuration interaction (FCI) calculations that are performed using classical computers. Full configuration interaction calculations solve the underlying quantum physics equations that govern the chemical interactions exactly, but are computationally very expensive and applicable to only the smallest molecules.
This proof of concept is a necessary benchmark for the new approach, as FCI provides the highest level of accuracy for chemical simulations. These preliminary findings—which are currently undergoing peer review—mark the largest molecular system ever simulated with such accuracy on a quantum computer.
“The successful demonstration of problem decomposition techniques on IonQ’s cutting edge hardware represents another step in Dow’s journey toward leveraging the enormous potential of quantum computing for innovation in the materials science and chemistry sector,” says Andre Argenton, Vice President of Core R&D, Dow. “This demonstrates Dow’s expertise as a leader in materials innovation together with 1QBit’s expertise in leading edge quantum computing software development.”
How Does it Work?
The method uses the principle of problem decomposition, which is a procedure that solves a complex quantum simulation problem by breaking it down into subproblems that are more compact and easier to solve.
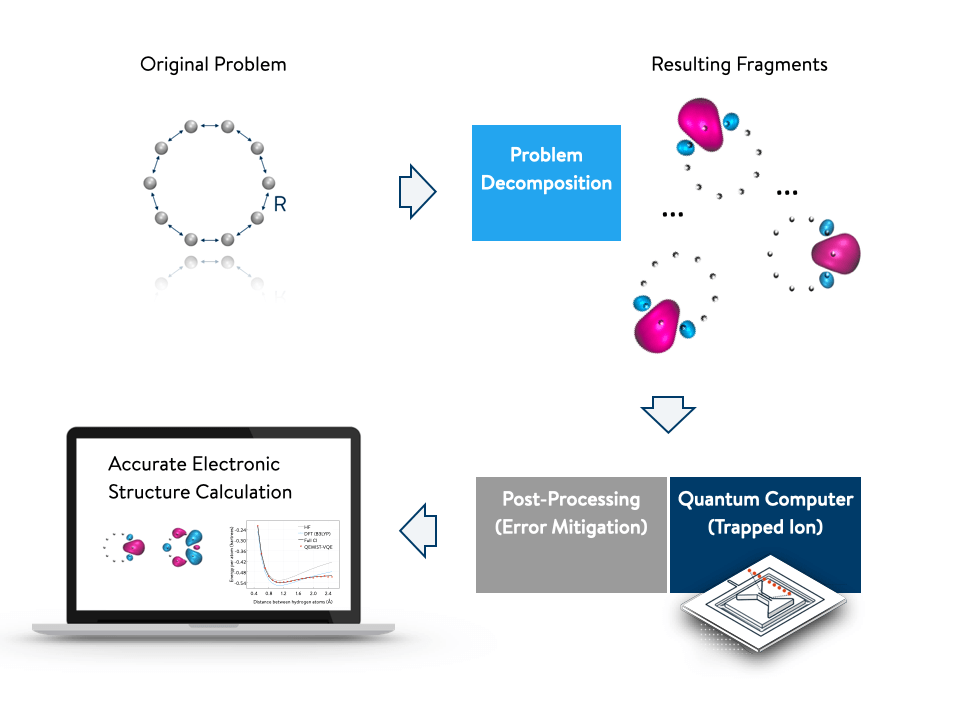
As a result, fewer qubits are required to achieve the same level of accuracy compared to solving the problem without decomposing it. A reduction in the required number of qubits can be as great as a factor of 10 while preserving accuracy.
Once the problem has been decomposed, the subproblems can be solved on a quantum computer, such as the trapped-ion quantum computer made available by IonQ. Then, a separate method efficiently mitigates the residual errors, further increasing accuracy. By combining several approaches in a single workflow, accurate electronic structure calculations may be performed from start to finish. The framework is also improvable with rapidly evolving quantum technology.
What Is the Outlook for Future Work?
Simulation of a typical, industrially relevant molecular compound could require more than 2000 qubits.2 Since the proposed framework can scale up with the rapidly evolving quantum technology, it can be used to significantly reduce qubit requirements for simulating such industrially relevant compounds, thereby extending the reach of quantum computation in the coming years. This approach provides a more practical framework for testing the potential of quantum computers to augment simulation of material properties.
For more details on this work, read the research paper.
References
¹ Y. Kawashima, M. P. Coons, Y. Nam, E. Lloyd, S. Matsuura, A. J. Garza, S. Johri, L. Huntington, V. Senicourt, A. O. Maksymov, J. H. V. Nguyen, J. Kim, N. Alidoust, A. Zaribafiyan, and T. Yamazaki, “Efficient and Accurate Electronic Structure Simulation Demonstrated on a Trapped-Ion Quantum Computer”, arXiv: 2102.07045, (2021).
2 P. Verma, L. Huntington, M. Coons, Y. Kawashima, T. Yamazaki, and A. Zaribafiyan, “Scaling Up Electronic Structure Calculations On Quantum Computers: The Frozen Natural Orbital Based Method of Increments”, arXiv: 2002.07901, (2020).