Microsoft and 1QBit Collaborate to Disrupt Materials Innovation with the 1QBit OpenQEMIST Platform and the Microsoft Quantum Development Kit
Innovation in materials design is important in a variety of industries, and computer simulation has been routinely used to help accelerate the materials-design process. Accurately predicting the chemical properties of a molecule at the quantum level is key to the design of new materials. But obtaining this information using classical hardware is computationally intensive. And the resources required for an exact solution scale exponentially with the size of the problem, quickly becoming intractable beyond the smallest molecules.
Because of this, accurate materials simulation has been limited to molecules that are relatively small. One of the biggest challenges in computational chemistry has been to scale materials simulation to the size of molecules relevant to industry, without compromising accuracy.
Quantum computers have the potential to simulate the quantum mechanical correlations of subatomic particles and accurately solve the electronic structure calculation problem using exponentially fewer resources than traditional methods. The shared quantum nature of programmable qubits and the systems being simulated allows for more efficient representations of the underlying physics. As such, performing electronic structure calculations is one of the most promising applications for near-term quantum computing devices. 1QBit has developed a solution that is compatible with the Microsoft Quantum Development Kit for this purpose.
QEMIST
1QBit has been developing a materials simulation platform called QEMIST, which stands for Quantum-Enabled Molecular ab Initio Simulation Toolkit. QEMIST is designed to enable the accurate calculation of molecular properties by leveraging the power of quantum computing in conjunction with advanced problem decomposition (PD) techniques.
The variety of PD techniques implemented in QEMIST enables massively parallel simulations by breaking down a computational chemistry task into smaller, independent subproblems. These subproblems can use a combination of interfaces to various classical and quantum solvers to achieve a higher level of accuracy for large-scale molecular simulations. QEMIST is developed primarily in Python, and its API supports seamless integration with the Microsoft Quantum Development Kit for the simulation of larger molecules.
OpenQEMIST
Today, 1QBit is releasing OpenQEMIST, providing access to a portion of the functionalities of QEMIST, as open source software under an Apache 2.0 license. This is aligned with Microsoft’s decision to open-source the Quantum Development Kit for the benefit of the rapidly growing community of quantum computing researchers and developers.
The 1QBit–Microsoft collaboration demonstrates how the integration of the two platforms expands the possibilities of quantum simulation. The samples below provide a glimpse into these possibilities.
The Microsoft and 1QBit Collaboration
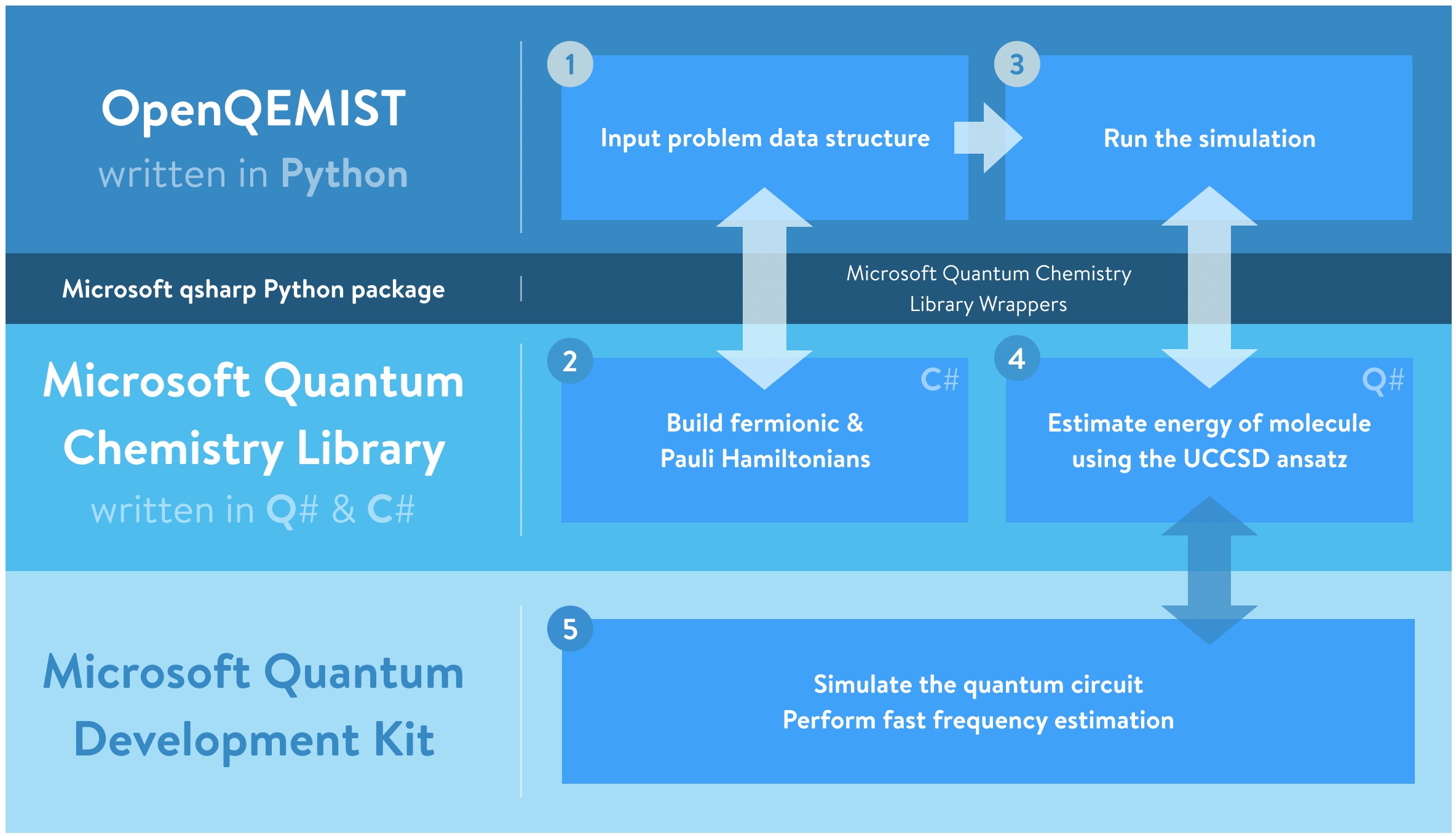 The Microsoft Quantum Development Kit provides a scalable end-to-end quantum development environment and leverages the Q# quantum programming language, which enables users to perform quantum algorithm design, compilation, and simulation. These functions are augmented by domain-specific libraries (such as the Microsoft Quantum Chemistry Library), and can be used in Python through the qsharp PyPI package. 1QBit has been working closely with the quantum team at Microsoft to connect the OpenQEMIST platform and the Quantum Development Kit using this package.
The Microsoft Quantum Development Kit provides a scalable end-to-end quantum development environment and leverages the Q# quantum programming language, which enables users to perform quantum algorithm design, compilation, and simulation. These functions are augmented by domain-specific libraries (such as the Microsoft Quantum Chemistry Library), and can be used in Python through the qsharp PyPI package. 1QBit has been working closely with the quantum team at Microsoft to connect the OpenQEMIST platform and the Quantum Development Kit using this package.
Together, 1QBit and Microsoft have developed an implementation of the variational quantum eigensolver (VQE) algorithm using Python for the classical components and interfacing with Q# to implement the quantum circuits. This implementation demonstrates the integration of the Quantum Development Kit with OpenQEMIST through the use, as an example, of density matrix embedding theory (DMET) for resource-efficient, larger-scale quantum simulations. The current release of Microsoft’s Quantum Chemistry Library includes key features that allow for the development and rapid testing of the VQE algorithm.
A Sample of Hybrid Quantum–Classical Computing
Currently, performing quantum chemistry simulations on near-term quantum computing platforms remains restricted to systems consisting of only a few atoms, due to the limited number of available qubits and gate operations, as well as the need for quantum error correction. Hybrid quantum–classical algorithms mitigate these restrictions by offloading well-suited computational tasks to existing classical approaches.
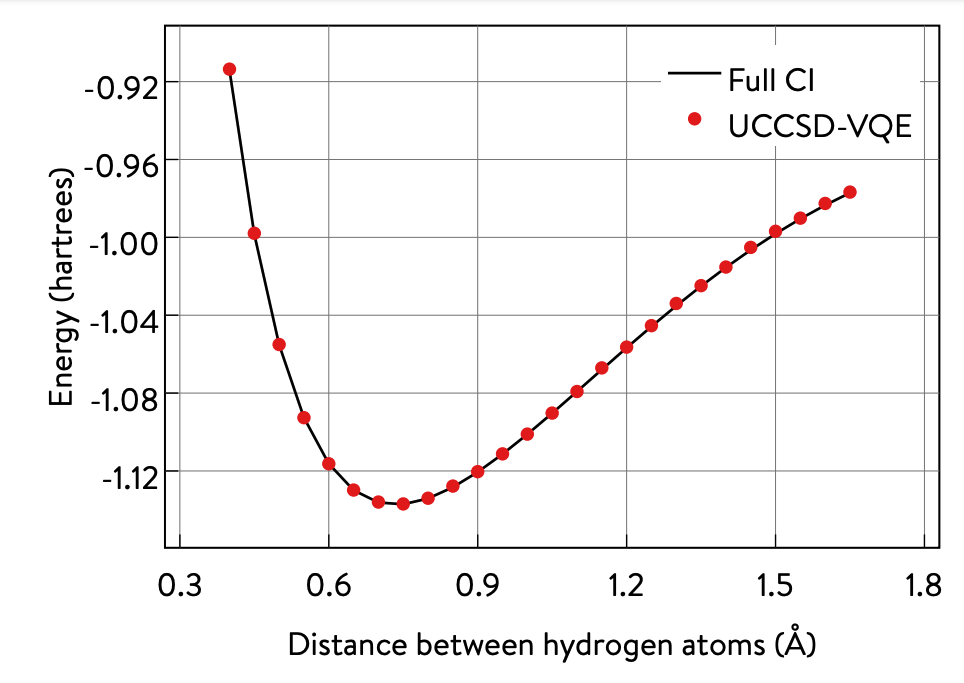
A bond dissociation curve of H2.
The recently developed implementation of the VQE algorithm demonstrates this combination of classical and quantum components. By combining quantum circuits with a classically controlled variational approach, it can perform electronic structure calculations while requiring a shallower circuit than other purely quantum algorithms.
A simple hydrogen bond dissociation experiment demonstrates the implementation of this hybrid algorithm. In addition, a detailed explanation and interactive sample of this hybrid approach is available, which also provides detailed information on how we used Python and Q# to implement this algorithm.
Problem Decomposition Techniques to Scale Up Hybrid Quantum–Classical Computing
After implementing the VQE algorithm, we can integrate this tool with QEMIST to leverage PD techniques and solve larger simulation problems. As mentioned, 1QBit has been exploring PD techniques that have the potential to scale up the size of molecules for quantum-enabled materials simulation by reducing the required quantum resources while maintaining the accuracy of the electronic structure calculation.
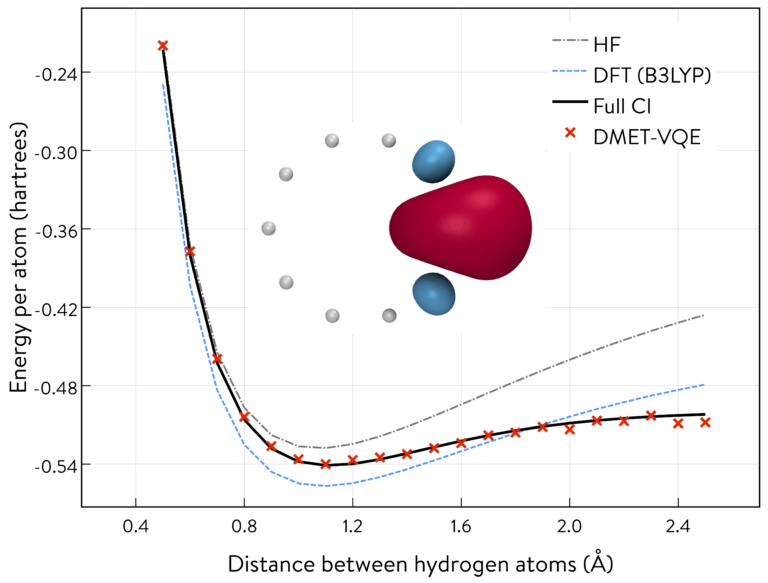
A potential energy curve of the symmetric stretching of a ring of 10 hydrogen atoms.
QEMIST is equipped with a variety of PD techniques and can intelligently select the most-appropriate techniques for a given molecular target. OpenQEMIST features DMET as a PD technique. It divides a molecule into fragments and determines the electronic structure of each subsystem using an accurate calculation method.
1QBit developed the DMET-VQE algorithm using the Microsoft Quantum Development Kit. It guides users through the construction of the potential energy curve of the “symmetric stretching” (i.e., changing the distance between the atoms homogeneously) of a ring of 10 hydrogen atoms.
The calculated results for DMET-VQE almost coincide with the Full CI calculation (i.e., the exact value) without using DMET on a classical computer. Running a VQE simulation of 10 hydrogen atoms without employing DMET requires 20 qubits with a minimal basis set. However, DMET-VQE uses only four qubits while maintaining the accuracy of the electronic structure calculation. Note that the other conventional quantum chemistry methods, the Hartree–Fock method (HF) and density functional theory (DFT) with the B3LYP functional, failed to reproduce the correct potential energy curve.
1QBit is a member of the Microsoft Quantum Network, and this successful demonstration of DMET-VQE, using OpenQEMIST and the Microsoft Quantum Development Kit, is a promising beginning to 1QBit and Microsoft’s collaboration efforts in materials design, paving the road toward practical quantum-enabled simulation to further accelerate progress in this area.
Resources for further exploration:
Most Recent News
How RBS Made a Quantum Leap into the Future
July 5, 2020 – The Sunday Times “In the shadow of a giant airship hangar on a defunct navy base in Silicon Valley, Ross McEwan sat down for a presentation about the future. It was 2015, and the then boss of Royal Bank of Scotland already had his hands full...
XrAI Trans Canadian Roll Out To Fight COVID-19
1QBit and Canadian health care providers team up to empower front-line clinicians with Health Canada’s first approved AI tool for radiology in the fight against COVID-19 Health and technology providers have joined forces to deploy XrAI, a machine learning tool that...
1QBit and Saskatchewan Health Authority Announce XrAI Chest Radiography Tool Deployment with Unprecedented Health Canada Regulatory Approval
Radiologist-created AI diagnostic product completes clinical trials, secures regulatory clearance, and goes live in response to the COVID-19 crisis. Vancouver, BC – April 8, 2020 – 1QBit, a world leader in advanced computing and software development, today announced...
Microsoft Partners with 1QBit, Honeywell, IonQ, and QCI on Azure Quantum
Today, Microsoft Quantum announced Azure Quantum, a full-stack, open cloud ecosystem that will bring the benefits of quantum computing to people and organizations around the world. The announcement highlighted the work done by partners such as 1QBit, Honeywell, IonQ,...
1QBit to Open Sherbrooke Office in Partnership with Université de Sherbrooke
Company expands to third quantum computing research hub in Canada Vancouver, BC – October 31, 2019 – 1QBit, a world leader in quantum computing software and hardware optimization, today announced that the company will be expanding its Canadian footprint by opening a...