The need to create new medicines faces major challenges due to the increasing costs of drug development and the high failure rate of drug candidates. These challenges are faced due to the laborious and expensive processes involved. For example, in order to develop an effective drug, it is necessary to test whether a drug candidate has sufficiently acceptable properties of ADME (absorption, distribution, metabolism, and excretion) and toxicity. Thus, there is a high demand for innovation and great opportunity for improvements in the drug discovery process.
In recent years, computational methods, such as virtual screening (VS), for identifying new drug candidates from an ever-increasing chemical library has helped to facilitate the drug discovery process. Computational VS can be understood as a kind of filter, where molecules are selected, according to particular criteria of potentially active molecules, against a specified pharmacological target. One of the VS approaches relies on the idea of identifying candidate ligands that are structurally similar to a known ligand that binds to the given target.

Molecular Similarity
Determining similarity between molecules is a practical endeavour due to the similar property principle from chemistry, which states that structurally similar molecules are expected to exhibit similar properties. That is to say, properties of molecules can be predicted by establishing and comparing their molecular structure. Molecular similarity measures can be categorized into two classes: graph based and fingerprint based.
Graph-Based and Fingerprint-Based Molecular Similarity Methods
Graph-based approaches are based on how one might intuitively conceptualize the molecular atom–bond structure, that is, with a mathematical graph, where nodes represent individual atoms or what are known as aromatic rings, and edges represent the chemical bonds between atoms. Each node and edge label can have assigned to them specific properties of the atom and bond, respectively. Moreover, the graph itself can have a label that carries some overall properties of the molecule.
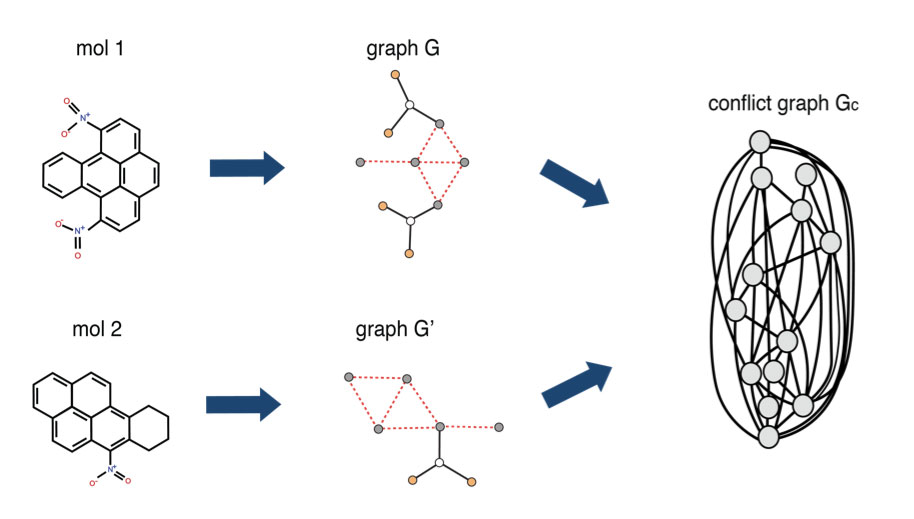
1QBit has developed a Graph-Based Molecular Similarity (GMS) method1 that compares two input graphs and returns two kinds of information: it provides a similarity score representing the degree to which the molecules have been judged to have similar structure as identified by the algorithm; and it identifies the structures they have in common. This method is formulated as a quadratic unconstrained binary optimization (QUBO) problem, which is a form suitable to be solved using a quantum annealer.
The GMS tool models molecules as graphs and builds a conflict graph. A conflict graph displays the structures common to the two parent graphs, where its nodes represent the potential matches and its edges represent potential conflicts in the selection of those nodes. The goal of the GMS tool is to find the largest set of vertices in the conflict graph that do not conflict, through the solving of an optimization problem. It then maps the solution to the original molecules to identify the common structure between the input molecules, and to quantify the molecules’ similarity score.
On the other hand, fingerprint-based measures are arrays of bits representing specific substructures in a molecule, where each bit is represented by either a one or a zero, indicating whether the molecule contains an associated substructure at a given level of probability. Although fingerprint-based methods are easy to use and computationally efficient, they cannot be used to assess whether a particular pattern is present in a molecular graph, as they do not usually consider the molecular structure.
Virtual Screening and the GMS Method
Virtual screening is a widely used strategy in modern drug discovery, and the GMS method2 is an valuable tool that can be applied to identify active compounds (i.e., the small molecules that can cause a direct physiological effect) in large datasets. 1QBit has implemented this novel GMS tool for the virtual screening of small molecules to assist in the finding of leading drug candidates in partnership with Accenture Labs and Biogen.
The GMS method provides more information about characteristics shared between molecules than traditional methods, which provide only a similarity score. Using this method, scientists and researchers can see exactly how and why molecular bonds match, which offers better insights and potential in helping to expedite the drug discovery process.

Key Benefits of the GMS Tool
Although the fingerprint-based method is able to compare molecules quickly, it does not offer information about the structural or chemical features that molecules have in common. The GMS tool can address this issue, as it identifies the common substructure between the molecules under comparison, affording valuable insights to the expert practitioner.
The study of small molecules and their properties plays a significant role in pharmaceutical research. For this reason, methods for the analysis and classification of small molecules based on their properties and functionality are critical to these research endeavours. Graph-based methods have shown great promise in practitioners’ ability to search, visualize, and infer new correlations between objects to identify, and thus possibly prevent, diseases, and to discover drugs customized to a given patient’s treatment.
Furthermore, improving the front end of the drug discovery process using quantum computing promises to dramatically cut costs and time to market, repurpose pre-approved drugs more easily for new applications, and help researchers and scientists to make discoveries much faster, which could lead to new cures for a range of diseases.
For more information on how the GMS tool can be used by pharma, read the following paper: A Quantum-Inspired Method for Three-Dimensional Ligand-Based Virtual Screening.
References
1 M. Hernandez, A. Zaribafiyan, M. Aramon, and M. Naghibi, “A Novel Graph-Based Approach for Determining Molecular Similarity”, arXiv:1601.06693 (2016).
2 M. Hernandez, G. L. Gan, K. Linvill, C. Dukatz, J. Feng, and G. Bhisetti, “A Quantum-Inspired Method for Three-Dimensional Ligand-Based Virtual Screening”, Journal of Chemical Information and Modeling, DOI:10.1021/acs.jcim.9b00195 (2019).